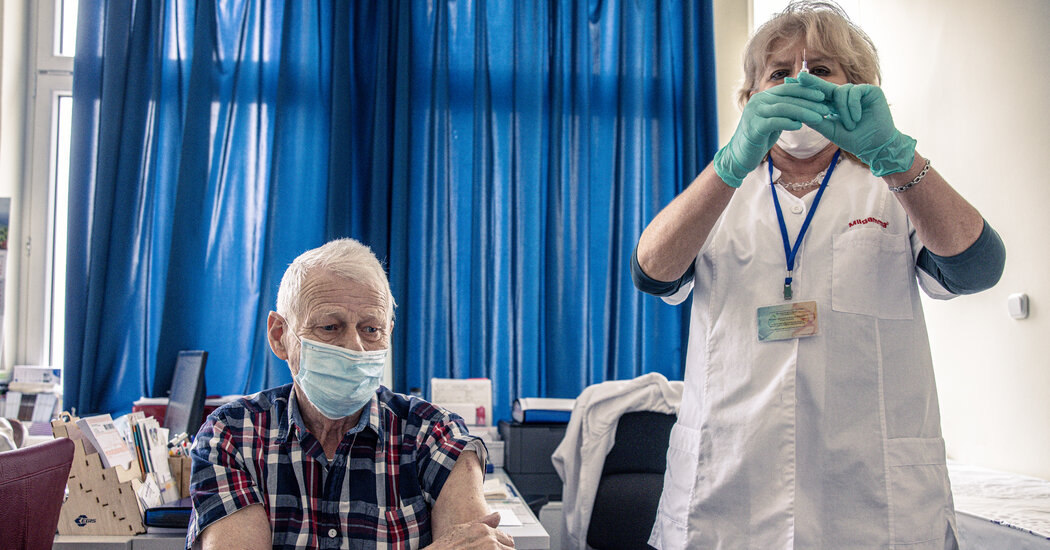
Hester Peirce, Commissioner for the U.S. Securities and Exchange Commission (SEC), Center, listens during a House Financial Services Committee hearing in Washington, DC, the United States, on Tuesday, September 24, 2019.
Andrew Harrer | Bloomberg | Getty Images
Hester Peirce is at a loss.
For years, the Securities and Exchange Commission, of which Peirce is a member, has rejected requests from national stock exchanges and financial companies to list securities that track the performance of the popular digital currency Bitcoin.
Back in the day – let’s say 10 years ago – concerns about possible market manipulation and liquidity might have made sense, but things have changed.
“This is probably the biggest and most frequently asked question I get: When will the SEC approve a Bitcoin publicly traded product?” Commissioner Peirce said in an interview with CNBC on Thursday.
“I thought if we had applied our standards as we applied them to other products, we would have already approved one or more of them,” she said. “With every day that goes by, the rationale we have used in the past for not being approved seems to be weakening.”
The SEC applies a “unique, elevated standard” to filings related to digital assets, it wrote in 2020. And it has argued that the agency is asking exchanges and potential ETF sponsors for assurances beyond what they do for traditional, stock-based demands products.
“People with a regulatory mindset say, ‘Oh wait, the market for Bitcoin looks a little different from the markets we’re used to,'” said Peirce on Thursday.
Now, she added, the Bitcoin market looks more like an established market with more institutional and established retail investors involved.
“So I think the markets have matured quite a bit,” said Peirce.
Renewed demands for a SEC-approved Bitcoin ETF come just weeks after the regulator announced its ruling on approving an application by VanEck to list shares of its Bitcoin Trust on the Chicago Board of Exchange’s BTZ Exchange move.
Regulators said in a letter dated June 16 that they would take additional time to seek comments from the public. In particular, the SEC asks investors and scientists for their opinion on whether Bitcoin ETFs could be susceptible to manipulation or whether Bitcoin itself is sufficiently distributed and therefore resistant to similar underhand manipulation.
But Peirce, a Republican named one of the SEC’s five commissioners by former President Donald Trump, has long denounced what she sees as double standards for Bitcoin products in her own agency.
Perhaps her sharpest objection came in a dissent in 2018, when she argued that the SEC should have approved an application by the Chicago Board of Exchange’s Bats BTZ Exchange to list and trade shares in the Winklevoss Bitcoin Trust.
“By excluding the approval of cryptocurrency-based ETPs for the foreseeable future, the Commission is operating a performance regulation,” she wrote at the time. “Bitcoin is a new phenomenon and its long-term viability is uncertain. It can be successful, it can fail. However, the commission is not well positioned to assess the likelihood of either outcome for Bitcoin or other assets. “
Three years later, VanEck’s current filing – much like pending Bitcoin ETF filings from Fidelity, Cathie Wood’s Ark Invest, and a few others – is viewed by the industry as an SEC litmus test now led by a cryptocurrency expert, Chairman Gary Gensler becomes.
Former chairman of the Commodity Futures Trading Commission, Gary Gensler, testifies at a US Senate Banking Committee hearing on systemic risk and market oversight on Capitol Hill in Washington on May 22, 2012.
Jonathan Ernst | Reuters
His appointment as head of the SEC by President Joe Biden, and his subsequent Senate confirmation, met with optimism from many in the crypto community as he is seen as a skilled hand in creating novel financial rules.
Gensler, who taught crypto courses at the Massachusetts Institute of Technology, is perhaps best known for his influential tenure as chairman of the Commodity Futures Trading Commission in the Obama administration. There Gensler helped develop and introduce a new supervisory system for the swap market, which was largely unregulated before the financial crisis.
Even if the Democrat Gensler does not necessarily agree with the Trump-appointed Peirce on all issues, they can join a more proactive SEC on Bitcoin regulation.
Rejecting Bitcoin ETF applications not only carries the risk of double standards, it can also provide few, more dangerous alternatives to thousands of investors.
“The Complications of Not Approval [an application] get stronger because people are looking for other ways to do the same things that they would do with an exchange traded product, “she said.” They are looking for other types of products that are not that easy to get in and out of maybe look at companies that are somehow related to bitcoin or crypto in a broader sense. “
Bitcoin itself has taken a violent start to summer and has fainted its price by more than 40% in the past three months. Although it remains one of the most actively traded digital assets, some market watchers say Bitcoin is at a critical juncture.
“It looks like it might be preparing for a $ 30,000 retest, and that could be critical,” Art Cashin, director of NYSE floor operations at UBS, said Thursday. “If you crack $ 30,000, traders will see if there is a trapdoor, a cascade sell-off, to follow.”
Zoom In Icon Arrows pointing outwards
Its dizzying ups and downs even come as a growing number of companies and banks, including payment companies Square and PayPal, began to facilitate Bitcoin transactions.
Meanwhile, the Bank of New York Mellon announced in February that it would start funding Bitcoin, a major development given that it is both the oldest bank in the country and a leader in custody banking.
Late on Friday morning, Bitcoin rose 1.6% to $ 33,550.
Despite the volatile fluctuations in the price of the currency, Peirce remains convinced that a Bitcoin ETF is overdue.
It is not the SEC’s job to approve or deny requests based on the merits of the investment itself, she said Thursday, especially if the exchanges meet legal requirements to protect investors from fraud.
“Bitcoin is so decentralized now. The number of nodes involved in Bitcoin is large and the number of people who have an interest in keeping this work decentralized is very large, ”she said. “People should make their own decisions: if people don’t want to buy bitcoin because they think it’s tampered with, they shouldn’t buy bitcoin.”